Contents
Contents
Click chemistry and nucleic acids
Click chemistry
Click chemistry is a chemical philosophy coined by Sharpless, which highlights the importance of using simple reaction conditions to join small molecular units together quickly and reliably, to ultimately imitate Nature’s methods of creating molecular diversity. A "click" reaction must
- be high yielding and stereospecific;
- be air- and moisture-insensitive;
- generate only inoffensive by-products than can be removed by non-chromatographic methods;
- be performed neat or in benign solvents.
Click reactions tend to have a high thermodynamic driving force, usually greater than 20 kcal mol−1. Such processes proceed rapidly and tend to be highly selective for a single product, i.e. "spring-loaded" for a single trajectory.
Copper-catalysed azide-alkyne cycloaddition reaction
1,3-Dipolar cycloadditions are a versatile approach for the preparation of a range of five-membered heterocycles. Under uncatalysed thermal conditions, the [3+2] cycloaddition of organic azides and alkynes normally produces two regioisomeric triazoles (Figure 1). In 2002, Meldal and Sharpless showed independently that copper(I) species catalyse the regioselective formation 1,4-disubstituted triazoles. In addition, copper(I) species have shown to increase the reaction rate by 107 with respect to the uncatalysed version.

Though there are other click reactions (such as alkene hydrothiolation reactions, nucleophilic subsitutions, epoxidations, dihydroxylations, aziridinations), the high yielding and regioselective copper-catalysed azide-alkyne cycloaddition has become synonymous with click chemistry, and has found a plethora of applications.
Catalytic systems
A number of different systems have been employed for the copper-catalysed azide-alkyne cycloaddition (CuAAC) reaction, including the use of copper(II) precursors with a reducing agent, copper(I) salts and pre-formed copper(I) complexes (Figure 2). Typically, the catalytic loadings for copper(II) salt/reducing agent systems tend be fairly high (up to 100 mol % copper and up to 10 equivalents of reducing agent with respect to copper). Large excesses of additives could affect other functionalities in the starting materials/products and increases the amount of waste and difficulty of isolating clean products.

Although these ligandless systems can be successful, they are limited to aqueous-based systems and hence water-tolerant substrates. Ligandless systems leave little room for improvement for unsuccessful CuAAC reactions. In sharp contrast, ligands can protect copper(I) centres, and have been shown to greatly enhance their activity and the rate of reaction. Pre-formed complexes are still scarcely used for this transformation, even though their use gives several advantages, most notably cleaner triazole products at lower copper loadings. Since copper(I) is cytotoxic even at low levels, these pre-formed complexes may find applications in biological systems.
Mechanism of the CuAAC reaction
Kinetic and DFT studies have shown that the reaction is not first order in copper (i.e. at least two copper species are required in the reaction). It is widely accepted that the reaction does proceed via a copper acetylide species, but the proposed catalytic cycle would begin with the formation of a copper-alkyne π complex (Figure 3). It is important to note that deprotonation is possible in aqueous media without the need for base as calculations have shown that copper coordination increases the acidity of the alkyne proton by up to 9.8 pH units. The organic azide can then coordinate to a copper centre in two different ways: an end-on fashion achieved by binding to the copper with the terminal nitrogen; or through the carbon-linked nitrogen. The mechanism would then proceed via a six-membered metallacycle transition state to the copper triazolide species, which would undergo reductive elimination to furnish the 1,4-disubstituted triazole and close the catalytic cycle.

Strain-promoted azide-alkyne cycloaddition (SPAAC) reaction
Without a copper(I) source, the cycloaddition reaction of organic azides with terminal alkynes is sluggish, however the cytotoxicity of copper can limit the applicability of this reaction in biological chemistry. In 1961, the reaction between neat cyclooctyne (the smallest stable cycloalkyne) and phenyl azide was reported to proceed "like an explosion" and gave a single product, the desired triazole. This enhanced reactivity can be attributed to the release of ring strain (nearly 18 kcal mol−1). Over 40 years later, Bertozzi developed the SPAAC reaction, which occurs under physiological conditions without any copper(I) source. It was later discovered that the addition of two fluorine atoms to cyclooctyne (Figure 4) can increase the rate of reaction but such alkynes suffer with some water-solubility issues, and require many steps for their preparation. The Boons group discovered that dibenzocyclooctynes exhibit enhanced reactivity towards azides. These alkynes are, in general, easier to prepare than fluorinated cyclooctynes and can be derivatised at various aryl positions to tune the reactivity and/or solubility. A phosphoramidite group has also been added to the cyclooctyne for insertion during solid-phase oligonucleotide synthesis.

DNA click chemistry
Click chemistry, and the azide-alkyne cycloaddition reaction, has, in a short space of time, become an important tool in nucleotide synthesis. Click chemistry is now used routinely in labelling DNA oligonucleotides with different functionalities, for example, biologically active moieties and fluorescent dyes; and click chemistry has recently found an exciting range of applications in the synthesis of longer oligonucleotides.
Click oligonucleotide labelling
The most common method of post-synthetic oligonucleotide labelling involves amide bond formation from an amino-modified and the N-hydrosuccinimide ester of a dye (Figure 5a). A disadvantage of this method is that N-hydrosuccinimide esters are readily hydrolysed in aqueous media.
The CuAAC reaction provides a method of labelling oligonucleotides with an array of functional groups, including chimeric molecules, epigenetic modifications and moieties that might be unstable to enzymatic ligation reactions (Figure 5b,c).

The CuAAC reaction is orthogonal to standard amino-link labelling, and usually gives high yields in product under mild reaction conditions. Unfortunately, there is a lack of commercially available dye-linked azides and alkynes. Safety must also be taken into consideration: azides are toxic and explosive substances that can decompose to release nitrogen gas through the input of energy, such as pressure, heat or impact. Ionic azides (such as sodium azide) are relatively stable, however low molecular weight and covalently-bound inorganic azides are particularly unstable. For organic azides to be non-explosive the general rule is that the number of nitrogen atoms must not exceed that of carbon, and that (NC + NO)/NN ≥ 3 (NC, NO and NN are the number of carbon, oxygen and nitrogen atoms, respectively).
Oligonucleotide ligation and cyclization
Although there has been great interest in the preparation of synthetic oligonucleotides over the last 30 years, the chemical synthesis of long strands of DNA (current limit around 150-200 bases) is plagued with problems such as low overall reaction yields, imperfect coupling efficiency and side reactions. Consequently, shorter sequences are usually subjected to multiple enzymatic ligation reactions, using large excesses of primers – an expensive process. The CuAAC reaction has been exploited for this application, since, not only is it a reliable reaction which can be performed using cheap, commercially available reagents, the azide and alkyne functionalities can be easily introduced.
Unlike the cycloaddition reaction with small, simple organic molecules, the azide-alkyne cycloaddition with DNA molecules normally requires a template to bring the cycloaddition partners in close proximity to improve the efficiency of the reaction. This template is simply a short length of DNA (called a splint), which has a complementary sequence in the region where the cycloaddition will take place (Figure 6).

Brown showed that a 300mer oligonucleotide (efficiently prepared from 3 100mer strands) could be used as a PCR template. In this instance, the entire sequence, including the bases on either side of the triazole linkage was copied correctly. Further studies have also shown that triazole-linked DNA can be biocompatible (Figure 7). Although the DNA triazole linkage on the left was read through by PCR, amplification of the resulting modified DNA template cause the loss of one nucleotide at the site of click ligation. This is possibly due to the presence of a rigid amide bond, which may cause the DNA strand to turn away from the growing DNA strand during replication. The linkage on the right better resembles a natural phosphodiester and is more flexible than the amide analogue, and is read through correctly during PCR.

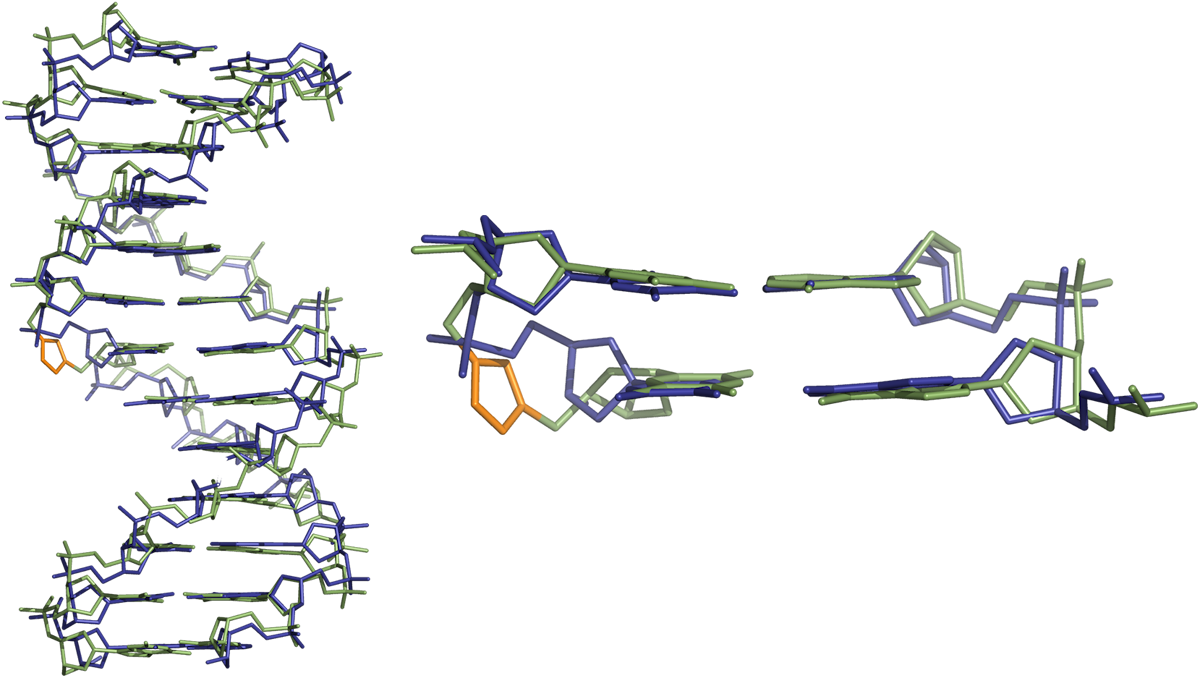
Whilst the change in the backbone induces locals changes in the helical parameters and reduces duplex stability (quantified by UV melting studies) the small change leaves the overall oligonucleotide structure intact, and the bases either side of the triazole linkage are able to form normal Watson-Crick base pairs (Figure 8).
Base and backbone analogues
The triazole ring has been used as a replacement for standard Watson-Crick bases. Unfortunately, none of the artificial nucleobases produced stable DNA duplexes with complementary strands. Interestingly, despite the low stability, oligonucleotides containing the (phenylthio)methyl azide derivative gave similar UV melting temperatures when paired with complementary strands with any of the four normal nucleobases (Figure 9). This suggests that it could be a condition as a universal base.

Unnatural triazole backbone
A number of DNA analogues that incorporate triazole units have been synthesised via the CuAAC reaction. For instance, Isobe and co-workers designed a new triazole-linked analogue of DNA (TLDNA), in which every phosphate unit was replaced by a triazole moiety using the CuAAC reaction (Figure 10). The preparation of this DNA strand was highly selective and efficient, and employs a completely different synthetic methodology, avoiding air- and moisture-sensitive phosphoramidite coupling chemistry and should be amenable to large-scale synthesis and/or automation. This promising analogue could also compete with PNA as an analogue to DNA, if the incorporation of all four Watson-Crick nucleobases can be perfected. One disadvantage may be that the product may not resemble or mimic normal, phosphate-containing DNA strands or may not be compatible with PCR.

Further reading
For an introduction to the Click philosophy, see: H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021.
Discovery of the CuAAC reaction: V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064.
Special issue on Click chemistry: Chem. Soc. Rev., 2010, 39, 1221–1408.
Overview of ligands/copper complexes used in the CuAAC reaction: S. Díez-González, Catal. Sci. Technol., 2011, 1, 166-178.
For further examples of strain-promoted cycloadditions using alkynes and other dipoles, see: C. S. McKay, J. Moran and J. P. Pezacki, Chem. Commun., 2010, 46, 931-933; F. L. van Delft, G.-J. Boons et. al., Angew. Chem., Int. Ed., 2010, 49, 3065-3068; G.-J. Boons et. al., J. Am. Chem. Soc., 2011, 133, 949-947.
N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046-15047.
X. Ning, J. Guo, M. A. Wolfert, G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253-2255.
A. H. El-Sagheer, A. Pia Sanzone, R. Gao, A. Tavassoli and T. Brown, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 11338-11343.
A. Pia Sanzone, A. H. El-Sagheer, T. Brown and A. Tavassoli, Nucleic Acids Res. 2012, 40, 10567-10575.
M. Shelbourne, X. Chen. T. Brown and A. H. El-Sagheer, Chem. Commun, 2011, 47, 6257-6259.
M. Nakahara, T. Kuboyama, Akhiro Izawa, Y. Hari, T. Imanishi and S. Obika, Bioorg. Med. Chem. Lett., 2009, 19, 3316-3319.